Research Interests
Outline: Molecular modeling package “GALAXY”
The ultimate goal of our research is to understand and to predict the functions of biomolecules in atomic details with theoretical and computational methods. Such research can also provide basic tools for the design of small molecules or proteins that can contribute to promoting human health.
For such purposes, we have been developing a biomolecular modeling program package GALAXY and a web server GalaxyWeb implementing the GALAXY methods1 (click here to connect to the web server).
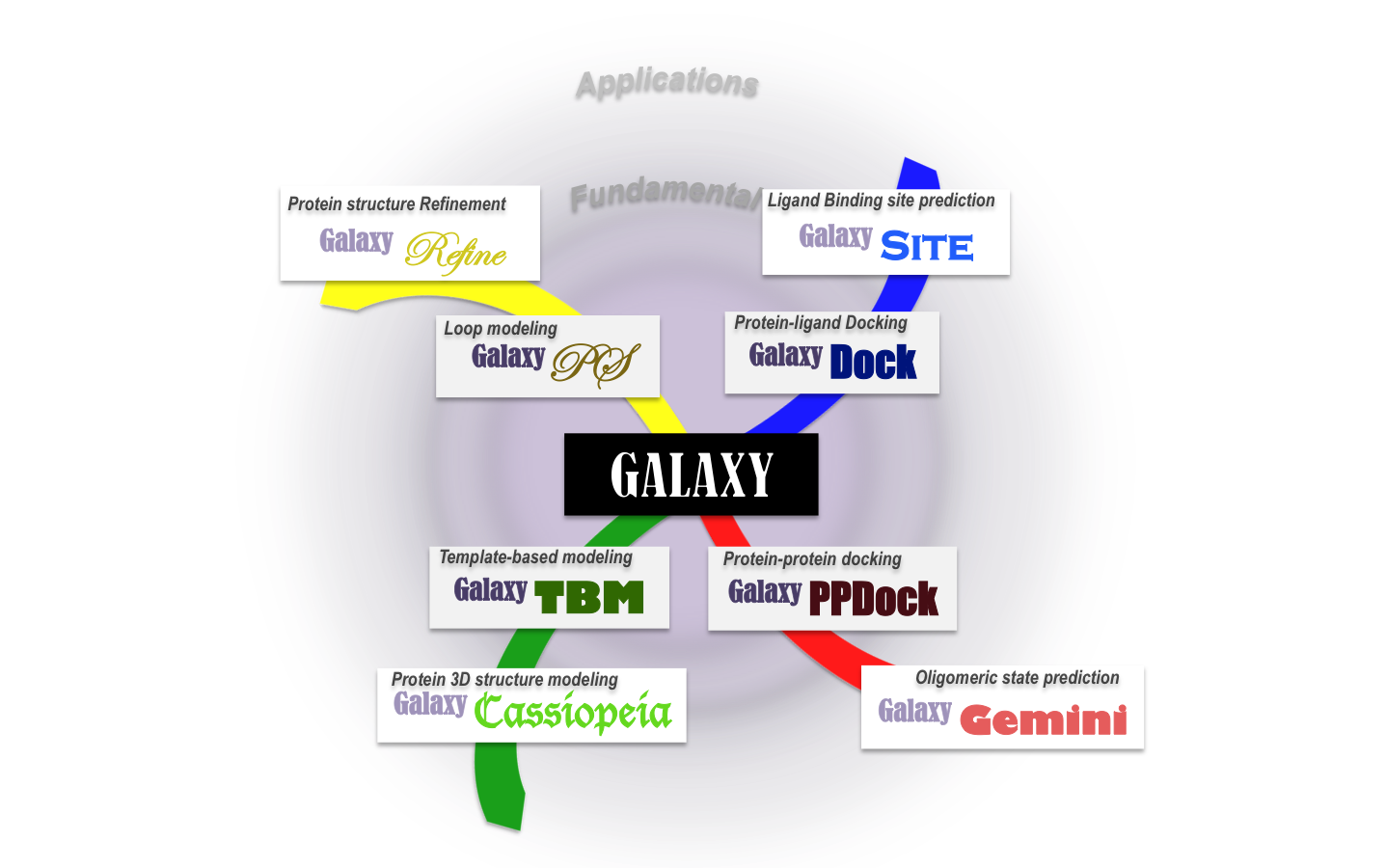
GALAXY is a software package for biomolecular modeling which consists of core programs that perform basic modeling tasks such as protein structure prediction, protein-ligand docking, and protein-protein docking and applied programs aimed at more specific goals such as ligand binding site prediction or protein homo-oligomeric state prediction. The GALAXY program package has been designed to be modular, enabling convenient addition of new programs in the future.
The philosophy underlying the GALAXY development is to provide more “practically useful” information to experimentalists, such as more “reliable” model structures, by fusion of available tools and knowledge in diverse disciplines such as chemistry, physics, computer science, or bioinformatics. GALAXY currently utilizes various scoring functions (e.g. molecular mechanics, implicit solvation models, statistical scores) and sampling techniques (e.g. molecular dynamics, genetic algorithm, Monte Carlo simulation, conformational space annealing) to achieve the best performance in each modeling task.
Protein Structure Prediction
Template-based modeling in CASP experiments
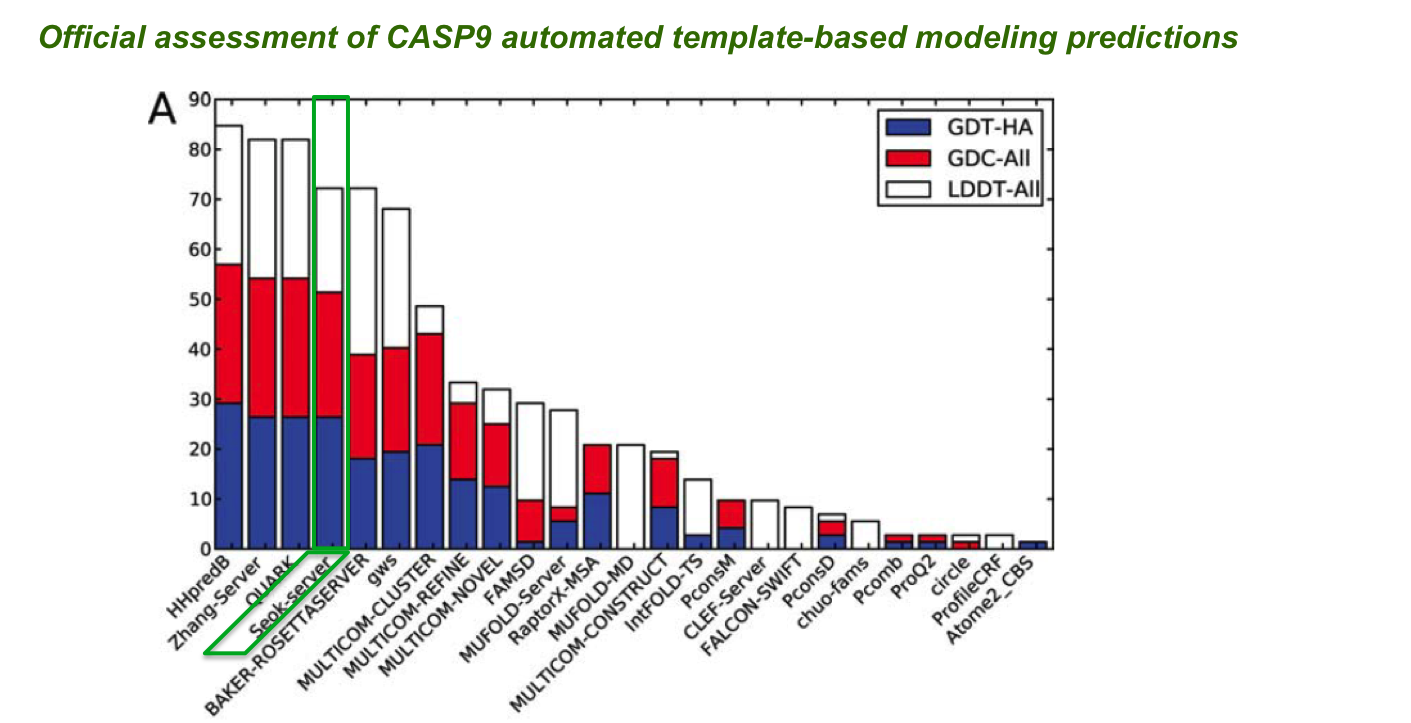
The CASP (Critical Assessment of the techniques for protein Structure Prediction) experiment is a community-wide protein structure prediction competition in which more than 100 groups from various countries test their prediction methods. In our first participation in CASP (CASP9, 2010), our automated prediction server “Seok-server” (based on the “GalaxyTBM” method2) was evaluated to be “among the top six servers” in the template-based modeling (TBM) category (the largest and the most important category in CASP) according to the official assessment by the CASP organizers.
GalaxyTBM: Automated protein structure prediction
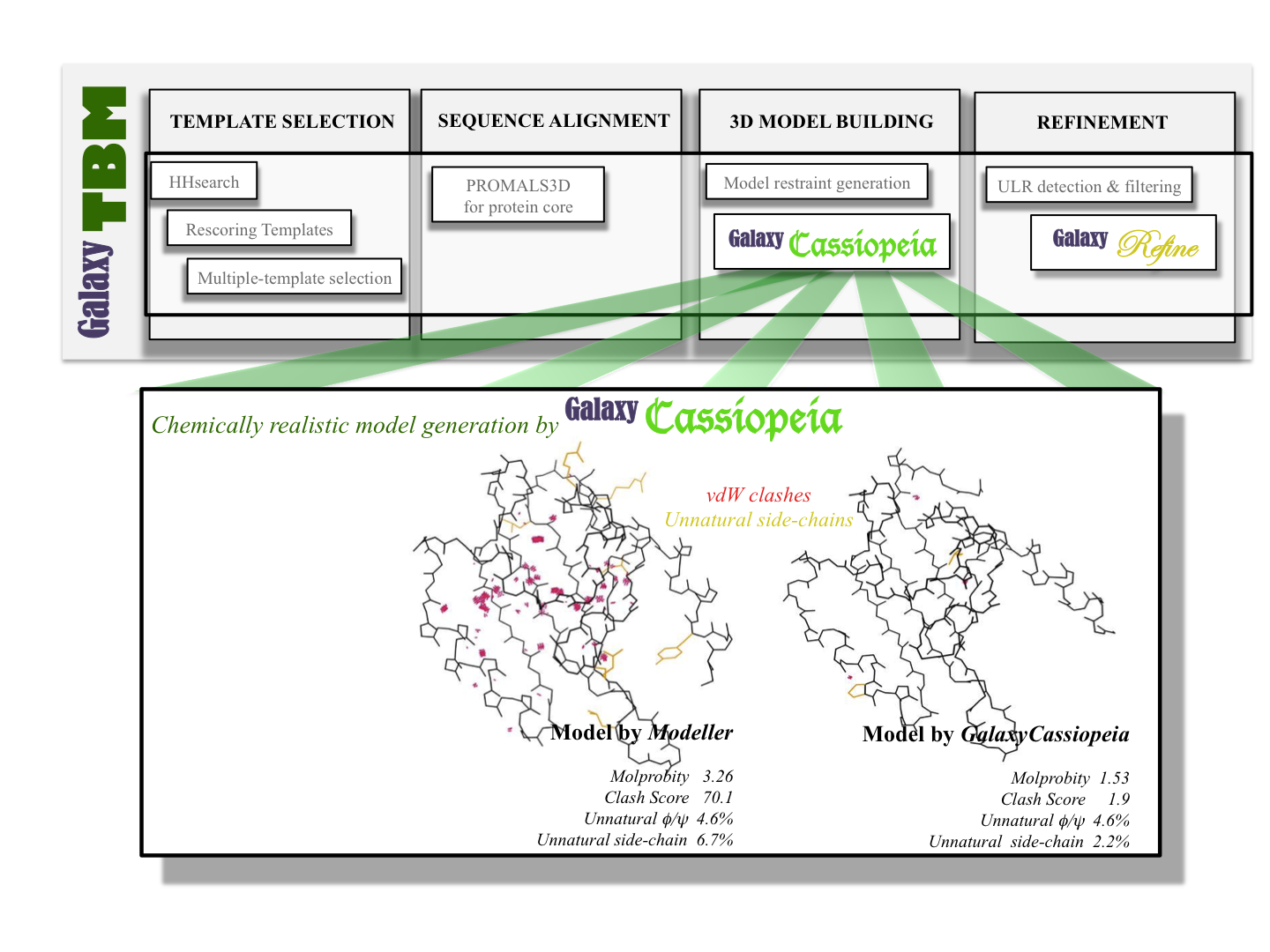
Our stand-alone tool for template-based modeling is called “GalaxyTBM” 2 (web
service available at GalaxyWeb).
GalaxyTBM combines a number of modules in the GALAXY package such as
GalaxyCassiopeia and GalaxyRefine and external programs for template detection
and sequence alignment.
One of the merits of GalaxyTBM is that it automatically refines unreliable model regions in relatively short computation time. Unreliable loops, termini, or side-chains are automatically detected and reconstructed by Galaxy modules. The generated model structures are not only globally accurate but also locally realistic. We keep improving the components of GalaxyTBM, both in the performance and speed, to provide better services to users.
Protein Loop Modeling
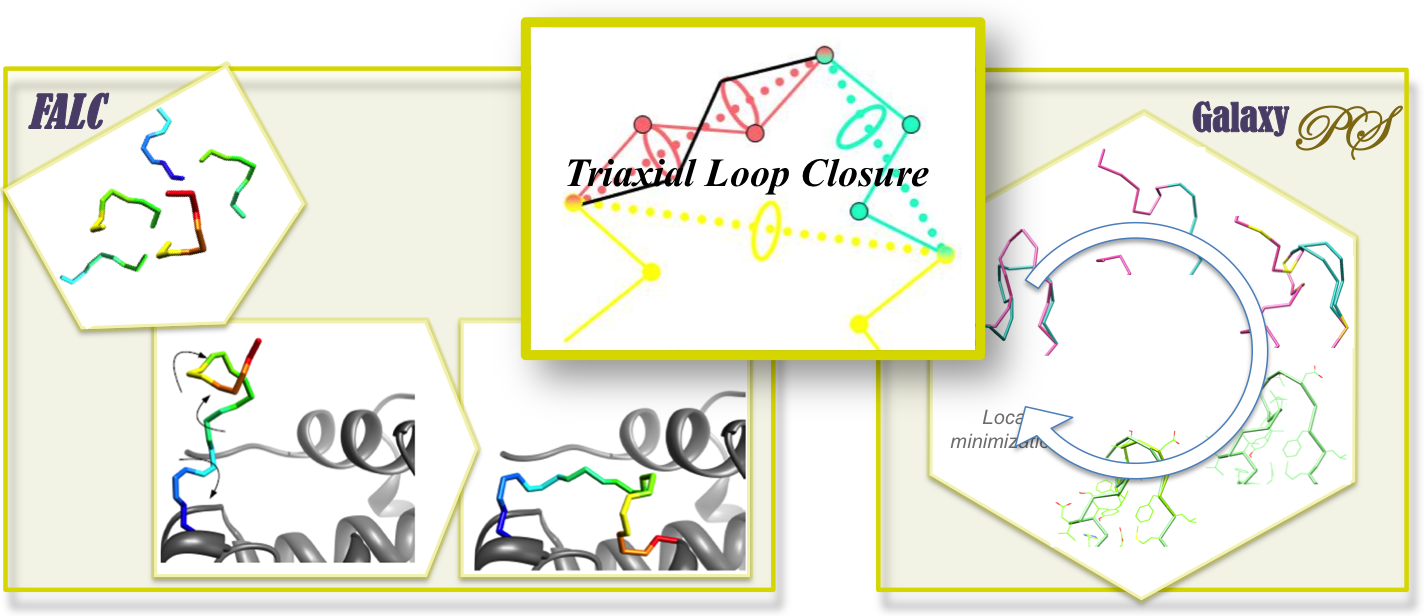
Protein loop modeling is one of the areas of our specialty. Loop modeling is applicable to the studies related to protein flexibility. All our loop modeling methods have grounds on the loop closure algorithm called “triaxial loop closure”34. For example, an ensemble of loop conformations can be generated by “FALC”56 that combines the loop closure and the local structure sampling technique of fragment assembly (web service available at FALC web server). In the more advanced loop modeling method “GalaxyPS”789 (web service available at GalaxyWeb, loop closure is used as an elementary move set during global energy optimization.
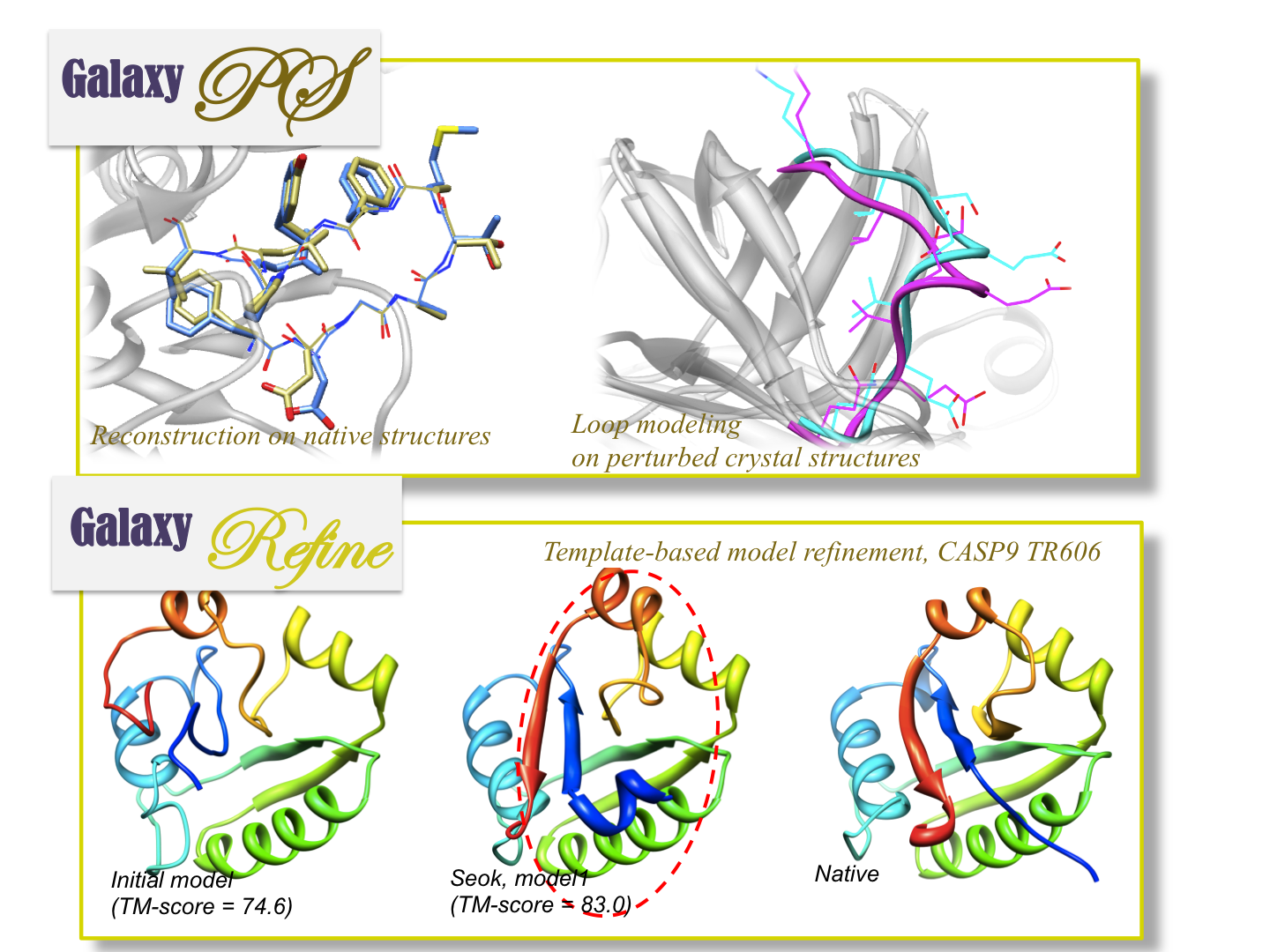
In contrast to traditional loop modeling studies that focus primarily on reconstructing loop structures in the experimentally resolved structures, we are extending “GalaxyPS” loop modeling to a broader range of problems. For example, even in perturbed crystal structures or structures in different functional state, GalaxyPS produces accurate predictions in the atomic level910. Therefore, this approach is expected to be more useful for solving more practical problems.
Loop modeling method can be further applied to improving template-based models. A template-based model refinement tool called “GalaxyRefine” was developed based on GalaxyPS. GalaxyRefine has been applied to CASP experiments since CASP9, and the model quality improvement by GalaxyRefine has been demonstrated in CASP9 TBM and refinement categories8.
Protein-Ligand Docking
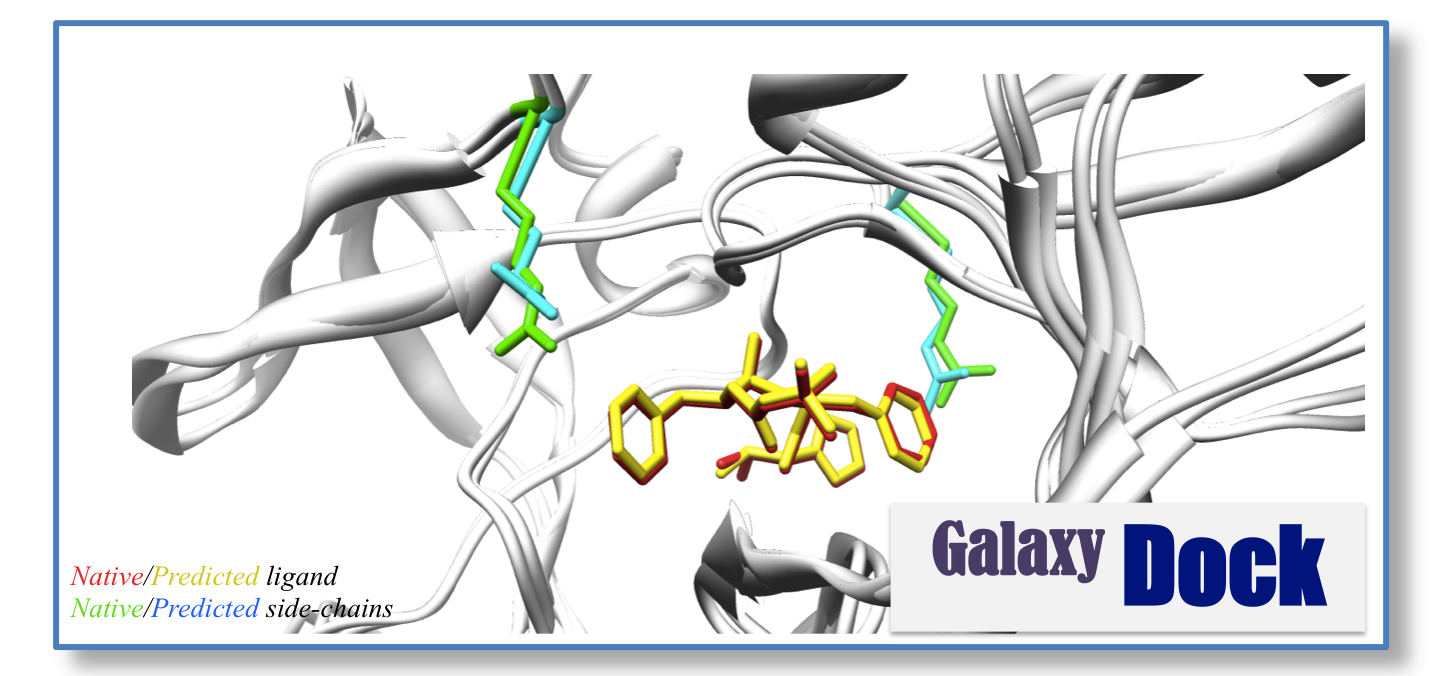
Another essential computational tool necessary for understanding protein function is protein-ligand docking. A protein-ligand docking method predicts the bound pose of the ligand and the binding affinity. Protein-ligand docking is one of the essential tools for computer-aided drug discovery.
Our recent research in this area focuses on incorporating receptor (protein) flexibility to our docking program “GalaxyDock”1112. A large number of proteins undergo conformational transitions to perform their biochemical functions. Nevertheless, many ligand-docking tools that treat proteins as rigid are used widely because of their efficiency and simplicity. A more practical docking program (with less computational cost and more accurate results) that considers receptor flexibility will bring a large impact to the field.
A related application method developed in our lab is the ligand binding site prediction method “GalaxySite”13. A key idea is to combine molecular docking and information from similar proteins. Molecular docking allows more precise prediction compared to other similarity-based methods. This method is accessible at GalaxyWeb.
Protein-Protein Docking
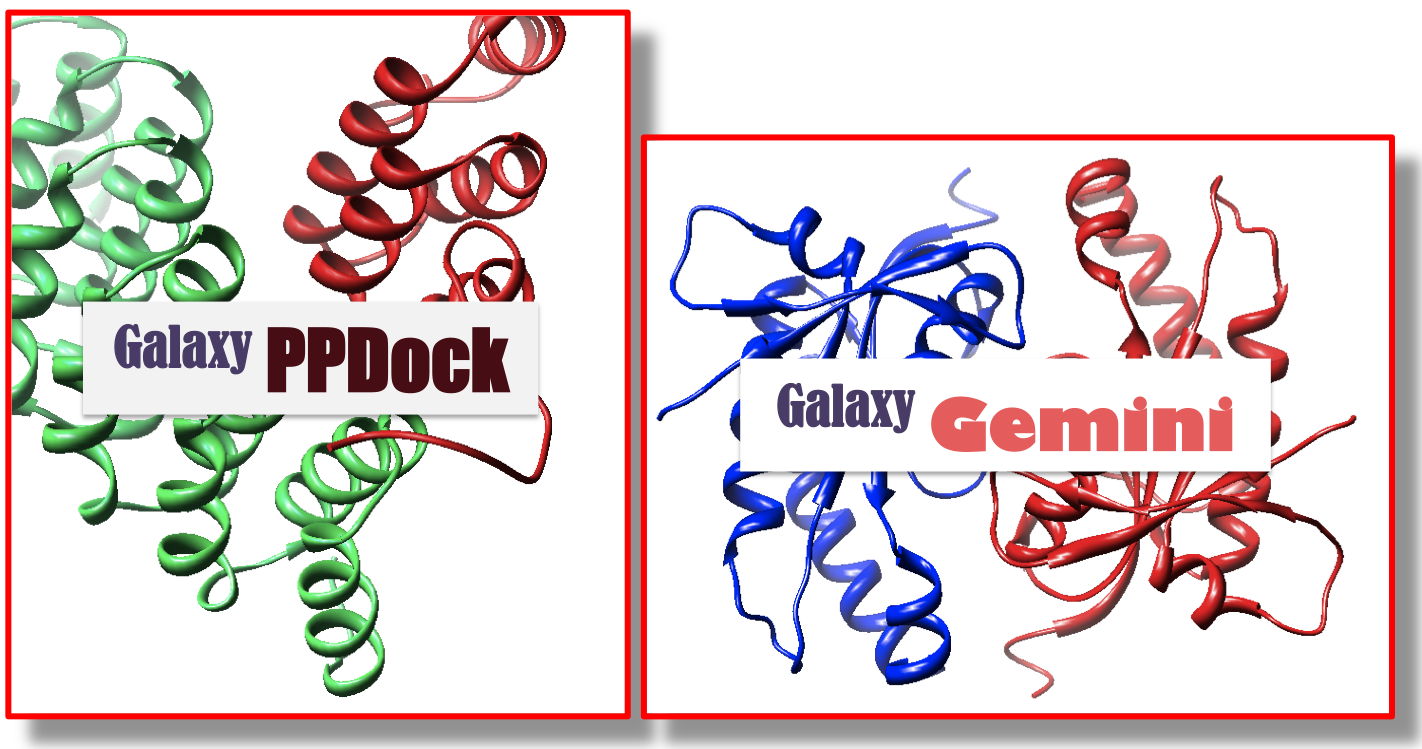
We are also developing prediction methods for protein-protein interactions. Prediction of protein- protein complex structure and binding affinity is crucial for understanding and regulating protein functions. Our protein-protein docking tool “GalaxyPPDock” is being tested in CAPRI14, a community- wide protein-protein interaction prediction experiment (Critical Assessment of PRedicted Interactions).
Understanding interaction between symmetric protein chains is a topic related to protein-protein docking. We developed a prediction method for homo-oligomer structure called “GalaxyGemini”15. GalaxyGemini makes use of known quaternary structures of template oligomers, selected based on sequence similarity and binding interface similarity to the target protein. Web service for GalaxyGemini is available at GalaxyWeb.
Applications
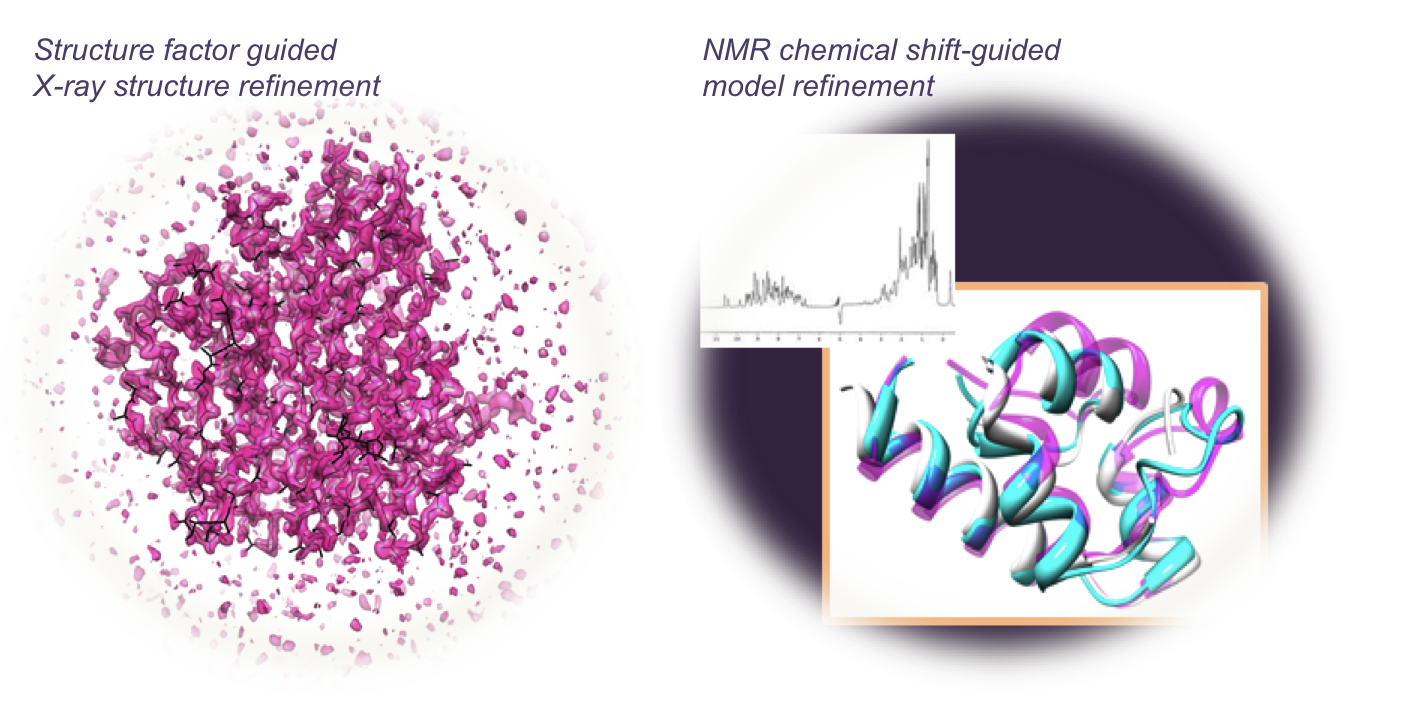
The next stage in the GALAXY development is to develop more tools for practical applications. The first type of application methods combine incomplete experimental data obtained by structural biologists (which may not be very useful as themselves) and structure prediction methods (which may not be accurate enough as themselves) to produce high-accuracy protein model structures.
For example, structure factors collected by X-ray crystallographers can be used to guide structure prediction even when phase information is not available and homology models are not accurate enough for molecular replacement. In addition, chemical shift data collected from NMR spectroscopy can assist refinement of template-based models. We are studying how to interpret and combine various experimental data with GalaxyTBM.
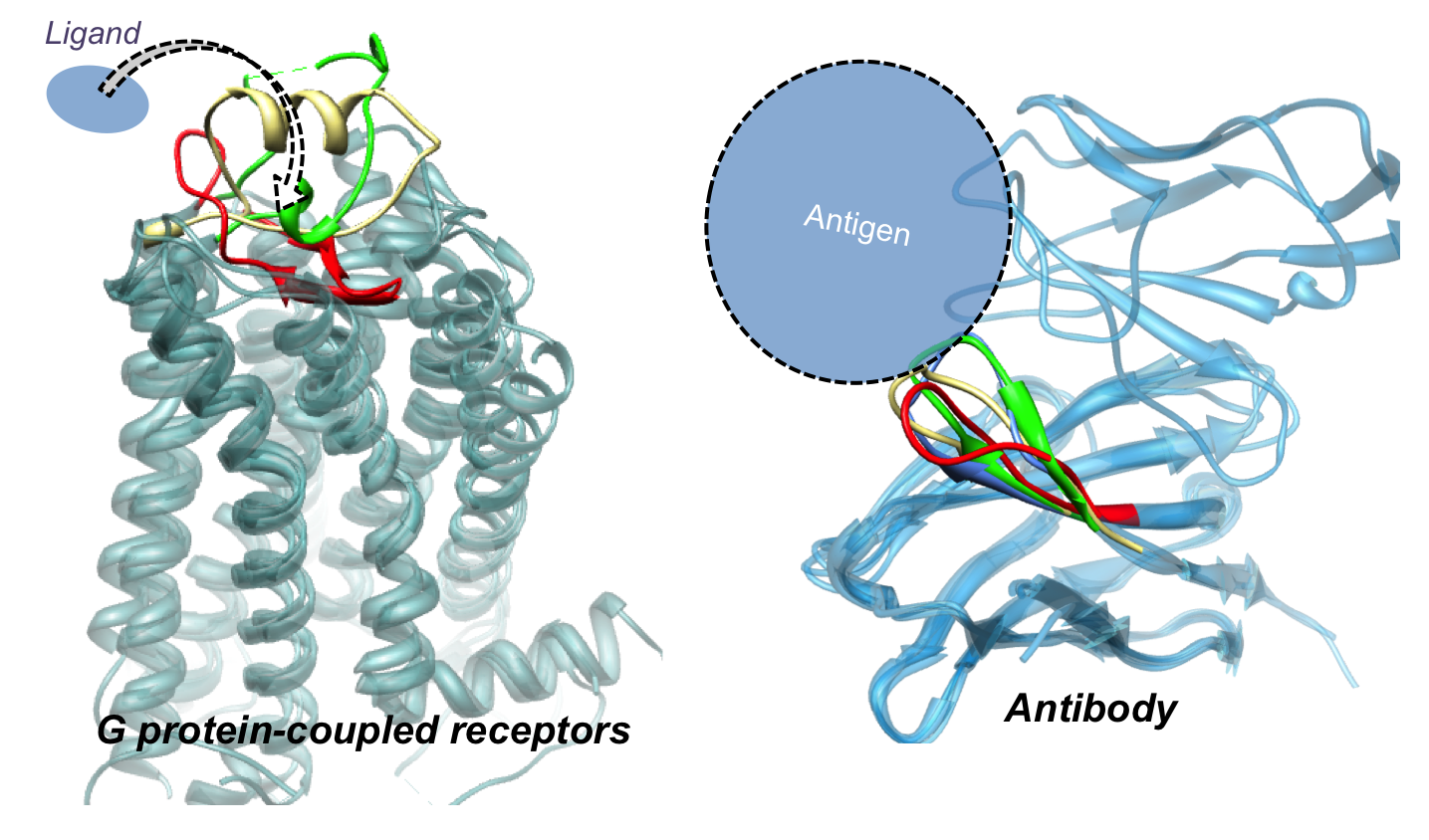
The second type of application methods aim at obtaining insights into conformational changes and molecular interactions involved in proteins of pharmaceutical interests such as antibody proteins and G protein-coupled receptors. Development of methods specialized for those targets by applying the current loop modeling and ligand docking techniques are undergoing, for example, modeling of intra- and extracellular loops of G protein-coupled receptors and modeling of antibody loop structures in the complementarity determining region.
References
-
J. Ko, H. Park, L. Heo, and C. Seok*, “GalaxyWEB server for protein structure prediction and refinement”, Nucleic Acids Res. 40 (W1), W294-W297 (2012). ↩
-
J. Ko, H. Park, and C. Seok*, “GalaxyTBM”, BMC Bioinformatics, 13, 198 (2012). ↩ ↩2
-
E. A. Coutsias, C. Seok*, M. P. Jacobson, and K. A. Dill, “A Kinematic View of Loop Closure”, J. Comput. Chem. 25, 510 (2004). ↩
-
E. A. Coutsias*, C. Seok, M. J. Wester, and K. A. Dill, “Resultants and Loop Closure”, Int. J. Quantum Chem. 106, 176 (2006). ↩
-
J. Lee*, D. Lee, H. Park, E. A. Coutsias, and C. Seok*, “Protein loop modeling by using fragment assembly and analytical loop closure”, Proteins, 78, 3428-3436 (2010). ↩
-
J. Ko, D. Lee, H. Park, E. A. Coutsias, J. Lee*, and C. Seok*, “The FALC-Loop web server for protein loop modeling”, Nucleic Acids Res. 39, W210-W214 (2011). ↩
-
H. Park, J. Ko, K. Joo, J. Lee, C. Seok*, and J. Lee*, “Refinement of protein termini in template-based modeling using conformational space annealing”, Proteins, 79, 2725-2734 (2011). ↩
-
H. Park and C. Seok*, “Refinement of unreliable local regions in template-based protein models”, Proteins, 80, 1974-1986 (2012). ↩ ↩2
-
G. R. Lee, W. Shin, H. Park, S. Shin, and C. Seok*, “Conformational sampling of flexible ligand-binding protein loops”, Bull. Korean Chem. Soc. 33 (3), 770-774 (2012). ↩
-
W. Shin, L. Heo, J. Lee, J. Ko, C. Seok*, and J. Lee*, “LigDockCSA: protein-ligand docking using conformational space annealing”, J. Comput. Chem. 32 (15), 3226-3232, (2011). ↩
-
W. Shin and C. Seok*, “GalaxyDock: Protein-ligand docking with flexible protein side-chains”, submitted. ↩
-
H. Lim, W. Shin, and C. Seok*, “GalaxySite: Ligand binding site prediction using molecular docking”, submitted. ↩
-
Sarel J Fleishman et al. (H. Park, J. Ko, H. Lee, C. Seok), “Community-wide assessment of protein-interface modeling suggests improvements to design methodology”, J. Mol. Biol. 414 (2), 289-302 (2011). ↩
-
H. Lee, H. Park, J. Ko, and C. Seok*, “GalaxyGemini: a web server for protein oligomer structure prediction based on similarity”, submitted. ↩
Funding
- BK21, Division of Chemistry and Molecular Engineering, SNU, KRF
- Basic Research Program, KOSEF
- Marine Biotechnology Program funded by Ministry of Land, Transport and Maritime Affairs